Methods
We used a variety of tools and languages throughout the many components of our project, including R, RStudio, Plink, Python, and GCTA (Genome-wide Complex Trait Analysis) command line tool to name a few.



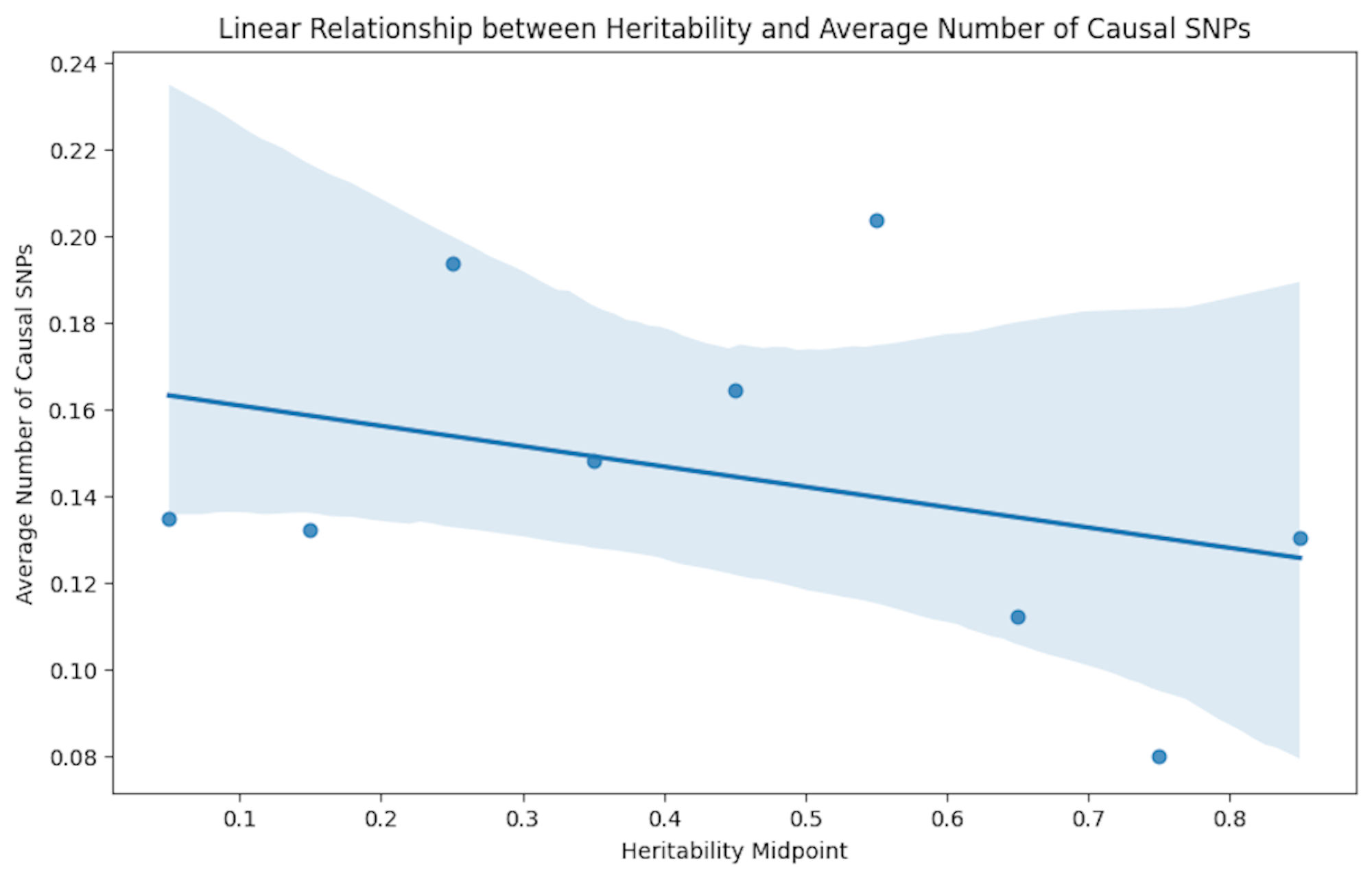
Regression
Illustrating the direct relationship between the causal variance and heritability.
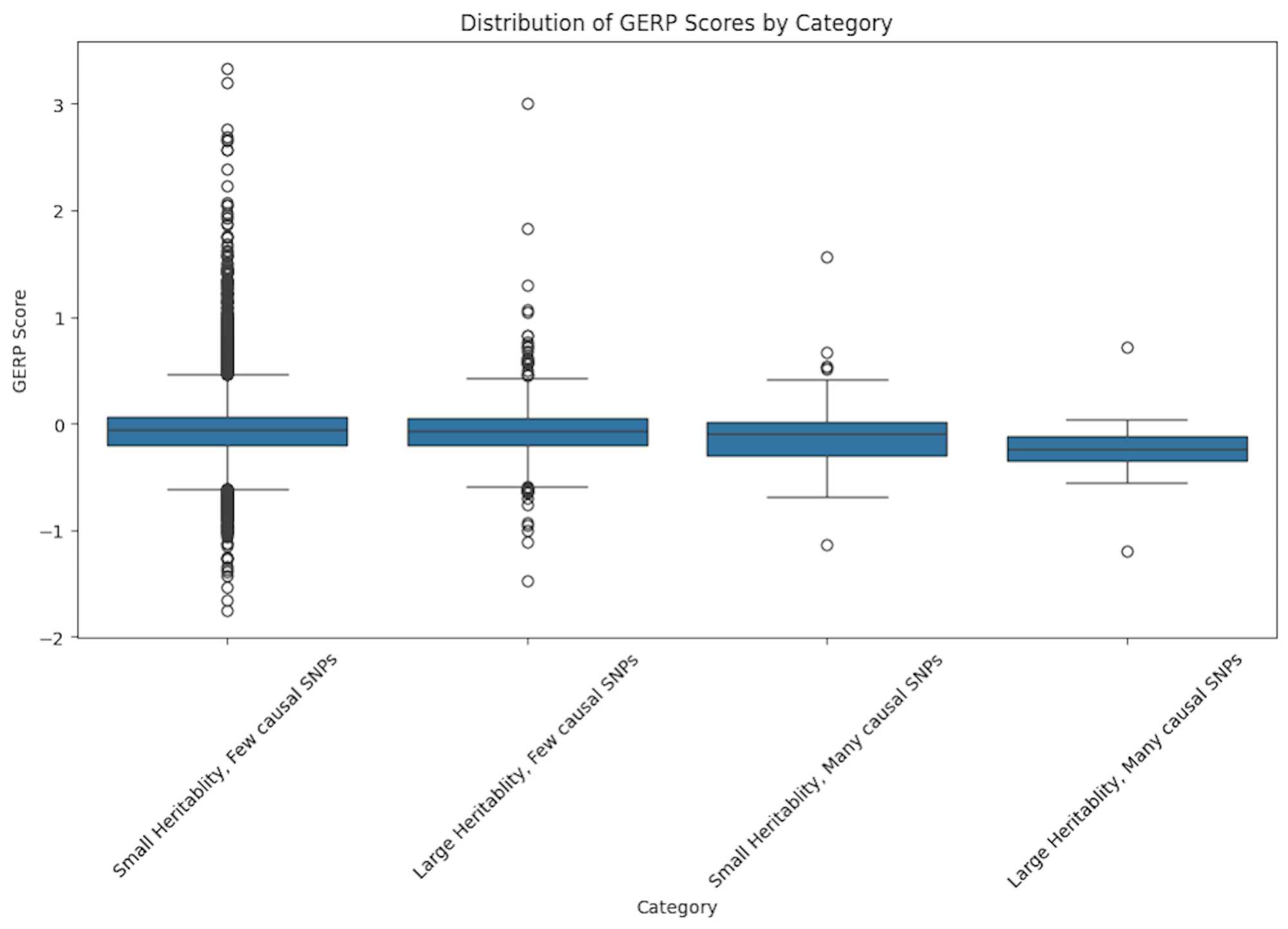
Genomic Evolutionary Rate Profiling (GERP)
The GERP score analysis offered additional evolutionary insight.
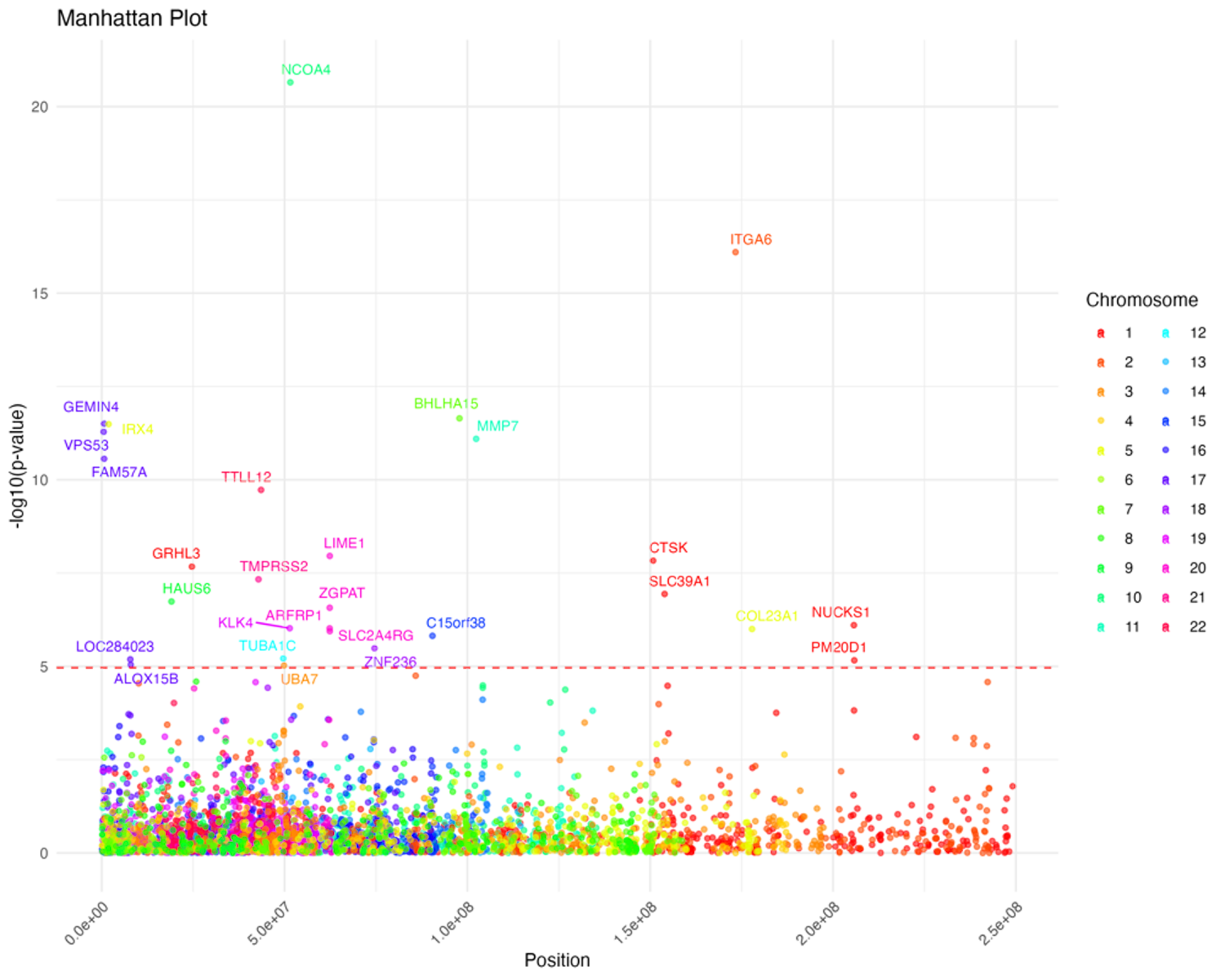
Transcriptome-Wide Association Study (TWAS)
The TWAS analysis is focused on cancer traits with high heritability and low causal variance to identify relevant gene IDs.
6 Models
TOP1 (Single best eQTL)The Sum of Single Effects (SuSiE) Fine mapping results
Bayesian Sparse Linear Mixed Model (BSLMM)
Elastic net regression
Least Absolute Shrinkage and Selection Operator (LASSO)
Best linear unbiased predictor (BLUP)
Note: This model was excluded from the results due to convergence issues